一、核心速览本文通过一系列密度泛函理论(DFT)计算,精准定位了三种分子材料单晶在弹性应力下能量储存的位置,深入揭示了分子材料弹性的起源,为柔性技术和智能设备的设计提供了关键理论依据。 从材料特性来看,研究聚焦于双(乙酰丙酮)铜(II)([Cu(acac)2],即[CuL21] )及其两种类似物[CuL22] 、[CuL23] 。[CuL21] 晶体在固态下具有显著的弹性柔韧性,能反复弯曲且不损失结晶度 。其晶体结构由沿 [010] 方向延伸的平面分子链构成,分子间存在 π - π 堆积、CH - π 和 CH - O 等相互作用 。在弹性变形过程中,不同的分子间相互作用在拉伸和压缩应变下发挥着不同作用。拉伸应变时,π - π 相互作用主导恢复力;压缩应变时,CH - π 和 CH - O 相互作用产生恢复力 。这种独特的相互作用机制使得材料在弹性变形时,能量在不同相互作用间重新分配,从而实现可逆的形状变化。 在应用潜力方面,该研究成果对先进材料设计意义重大。明确分子材料弹性的原子尺度机制,有助于研发新型柔性建筑材料,使其在承受外力时能更好地适应变形且保持结构完整性 。在智能设备领域,如可穿戴设备、柔性显示屏等,能为材料选择和结构设计提供理论指导,提高设备的柔韧性和耐用性 。不过目前研究主要基于理论计算,未来需结合更多实验手段进一步验证和完善理论模型,同时探索更多具有特殊弹性性能的分子材料,拓展其在实际工程中的应用范围。 二、研究背景(一)弹性的重要性与研究现状弹性是一种广泛存在且至关重要的性质,无论是在生命科学还是工程技术领域都发挥着关键作用 。在生命体系中,弹性是细胞活动和动物运动的基础 。例如,肌肉的弹性收缩和舒张使得动物能够进行各种运动;细胞的弹性则影响着细胞的形态维持、物质运输以及与外界环境的相互作用 。在技术领域,弹性支撑着从光学纤维到摩天大楼等众多设计与建造 。光学纤维的弹性使其能够在弯曲和拉伸过程中保持光信号的稳定传输;建筑结构材料的弹性则确保建筑物在承受风荷载、地震等外力作用时,能够发生可逆变形而不致倒塌 。 尽管弹性在众多工程应用中具有不可替代的作用,并且人类对其应用已有数千年历史,但弹性恢复力的分子起源却一直未被完全理解 。在分子晶体中,弹性变形时共价键的变形可忽略不计,弹性行为主要由弱相互作用主导 。然而,目前人们通常将恢复力视为材料的宏观性质,认为是多种相互作用共同作用的结果,对于这些相互作用在原子尺度上如何协同产生弹性恢复力,仍缺乏深入了解 。这一知识缺口限制了依赖弹性的先进技术的发展,如柔性电子器件、智能材料等领域的创新。 (二)分子晶体弹性研究的机遇近年来,高精度测定弹性变形单晶的分子晶体结构技术取得了进展,为研究弹性材料中恢复力的原子起源提供了新契机 。通过这些结构信息,结合理论计算方法,能够深入探究分子间相互作用在弹性变形过程中的变化规律,从而确定恢复力的具体来源 。这不仅有助于从本质上理解分子晶体的弹性行为,还为设计具有特定弹性性能的新材料提供了可能 。例如,在设计新型柔性电子材料时,可以根据弹性起源的理论,精准调控分子间相互作用,优化材料的弹性和电学性能,满足不同应用场景的需求 。 (三)本研究的目标本研究旨在利用明确的弹性变形机制信息,通过理论计算深入了解晶体在受力时的能量变化,精确确定弹性恢复力的位置和使晶体恢复原始形状的机制 。以[CuL21] 为主要研究对象,分析其在弹性变形过程中分子间相互作用的能量变化,进而扩展到对[CuL22] 和[CuL23] 的研究,揭示不同分子结构对弹性行为的影响,为新型弹性材料的设计提供理论基础 。 三、研究方法(一)理论计算方法研究采用了多种理论计算方法来深入探究分子材料的弹性起源。首先,利用 CrystalExplorer 17.5 软件在 CE - B3LYP/6 - 31G (d,p) 水平上计算相互作用能、晶格能和能量框架 。这种方法能够对分子晶体中不同分子间相互作用的能量进行量化分析,确定主要相互作用类型及其相对强度 。例如,通过该计算发现[CuL21] 晶体中存在 π - 堆积(能量约为−80 kJ mol−1 )、CH - π(能量比 π - 堆积弱约−13 kJ mol−1 )和 CH - O(能量约为−10 kJ mol−1 )三种重要相互作用 。 对于[CuL21] 分子的计算,运用密度泛函理论(DFT)在无限制形式下进行,采用 ωB97xD 泛函研究固态下二聚体对的分子间能量 。计算中,C、H 和 O 原子使用分裂价三重 ζ 基组(6 - 311++G (d,p)),铜原子使用双 ζ 电化学势基组(LanL2DZ) 。为了修正基组叠加误差,采用了 counterpoise 方法,并在所有计算中应用了超细网格 。利用这些参数设置,能够更准确地模拟分子间相互作用在不同变形程度下的能量变化 。 此外,为了验证通过二聚体对建模晶体能量的方法,使用 VASP 软件,采用 B3LYP 泛函结合 DFT - D3 方法和 Becke - Johnson 阻尼函数对整个晶体结构的能量进行计算 。计算时,平面波展开的截止能量设置为 500eV,能量收敛标准为10−6eV ,在 Γ 点对布里渊区进行采样,并且在优化结构时保持晶胞形状和体积固定 。 (二)结构处理与分析从文献中获取晶体结构后,使用 SHELXL 软件对其进行修正,将氢原子固定在几何最优位置 。通过 Python 脚本对结构进行外推,生成在各种变形程度下的结构,包括超出材料测量弹性极限(约 1% 应变)的假设结构 。这些外推结构为研究不同应变条件下分子间相互作用的变化提供了基础 。利用这些结构数据,结合上述理论计算方法,能够系统地分析分子间相互作用能量随应变的变化规律,从而确定弹性恢复力的来源和机制 。 四、实验设计(一)分子材料选择选择了三种化学结构相似的分子材料:[CuL21] 、[CuL22] 和[CuL23] 作为研究对象 。[CuL21] (双(乙酰丙酮)铜(II))在晶体状态下具有明显的弹性柔韧性,已有的研究表明其晶体在弯曲过程中分子会发生旋转和滑移,导致晶体边缘的膨胀和压缩 。[CuL22] 和[CuL23] 分别是[CuL21] 中甲基被乙基和丙基取代的产物,它们具有不同的弯曲机制 。这种结构上的相似性和差异性,使得研究人员可以通过对比研究,分析分子结构变化对弹性性能和分子间相互作用的影响,从而更深入地理解分子材料弹性的起源 。 (二)计算内容设计针对选定的分子材料,计算内容主要围绕分子间相互作用能量在弹性变形过程中的变化 。对于每种材料,首先确定其晶体结构中的主要分子间相互作用类型,如[CuL21] 中的 π - π、CH - π 和 CH - O 相互作用 。然后,通过线性外推生成一系列不同变形程度的晶体结构,利用 DFT 计算这些结构中各种相互作用的能量变化 。在计算过程中,分别考虑拉伸应变和压缩应变两种情况,观察不同相互作用在不同应变条件下能量的增减情况 。同时,计算整个晶体结构的能量,与分子间相互作用能量之和进行对比,分析不同应变下两者的差异,以确定在弹性变形过程中起主导作用的相互作用以及其他可能影响弹性行为的因素 。 (三)对比验证设计为了验证计算方法的可靠性和结论的普遍性,进行了多方面的对比验证 。在计算方法上,对[CuL21] 采用不同的泛函(ωB97xD 和 B3LYP)和计算软件(Gaussian16 和 VASP)分别计算分子间相互作用能量和晶体结构能量,对比结果以确保计算的准确性 。对于不同的分子材料[CuL21] 、[CuL22] 和[CuL23] ,对比它们在相似应变条件下分子间相互作用能量变化的规律,分析结构差异对弹性行为的影响 。此外,还将理论计算结果与已有的实验数据进行对比,如晶体的弹性极限、弯曲机制等方面的实验观察,从不同角度验证研究结果的正确性 。 五、结果与分析(一)分子间相互作用的分析通过能量框架计算,确定了[CuL21] 晶体结构中的三种重要相互作用:沿 [010] 方向的链内 π - 堆积、通过 n 滑移面相关的相邻分子间的 CH - π(沿 [101] 方向分子间)和 CH - O(沿 [10] 方向分子间)相互作用(图 2) 。π - 堆积是面 - 面 π - π 和 Cu - π 相互作用的组合,能量约为−80 kJ mol−1 ,是相对较强的相互作用 。CH - π 可描述为相邻分子的 γ - 质子(H (3))与 γ - 碳(C (3))之间的边 - 面相互作用,比 π - 堆积弱得多,能量差约为−13 kJ mol−1 。CH - O 是最弱的相互作用,约为−10 kJ mol−1 ,由甲基(C (5))质子与氧原子(O (1) 和 O (2))之间的相互作用构成 。 对这三种相互作用进行 Morse 势拟合(图 3a - c),结果显示 π - π 相互作用的势阱深度为65.9 kJ mol−1 ,弹簧常数为22.9 N m−1 ;CH - π 相互作用的弹簧常数为25.9 N m−1 ,势阱深度为10.6 kJ mol−1 ;CH - O 相互作用的弹簧常数为24.0 N m−1 ,势阱深度为7.8 kJ mol−1 。这表明 CH - π 相互作用相对于 CH - O 相互作用,在距离变化时更难被拉伸或压缩,且势阱更深 。这种相互作用强度和特性的差异,对[CuL21] 晶体的弹性行为有着重要影响 。例如,之前的研究报道显示,晶体 [101] 面(CH - π 相互作用方向)的弹性模量较小,在弯曲晶体时比(10)方向(CH - O 相互作用方向)发生更大的变形,这与本次计算得到的相互作用特性结果一致 。 对于[CuL22] ,其相互作用的势阱深度与[CuL21] 量级相似,但 π - π 势更窄(势阱深度88.9 kJ mol−1 ,弹簧常数1.75Nm−1 ) 。而[CuL23] 则表现得更加刚性,弹簧常数大于600 N m−1 。这说明分子结构中引入不同长度的脂肪链会改变分子间相互作用的特性,进而影响材料的机械柔韧性 。脂肪链的引入增加了 CH - HC 相互作用位点,而这些相互作用通常是排斥的,使得材料的柔韧性降低 。 (二)弹性区域内相互作用的协同与竞争研究发现,[CuL21] 中各相互作用的最小值位于晶体弹性极限之外(π - π、CH - π 和 CH - O 相互作用分别在−2.1% 、4.2% 和11.4% 应变处) ,且材料整体的弹性区域远小于单个相互作用的弹性范围 。尽管 CH - π 和 CH - O 相互作用相较于 π - π 相互作用较弱,但它们在决定[CuL21] 的结构和弹性相互作用中起着关键作用 。由于在最低总能量结构中,没有一种相互作用达到最小化,所以[CuL21] 中的弹性相互作用存在 “受挫” 现象 。这种受挫的弹性相互作用类似于量子磁性和自旋交叉材料中的情况,在那些材料中,这种相互作用会导致一系列奇特的现象,并使系统状态具有 “柔软性” ,这可能与[CuL21] 的柔韧性相关 。 在弹性区域内,通过对分子和晶体计算的对比发现,三种主要相互作用(π - π、CH - π 和 CH - O)在小应变下主导了弹性能 。分子间相互作用能量之和与整个晶体结构计算能量的最小值都出现在未变形的晶体结构处,这意味着在弹性区域内,其他成对相互作用的影响较小 。然而,在较大应变下,分子间相互作用能量之和与晶体结构计算能量出现偏差 。这是因为在高应变下,即使单个二聚体的相互作用变得更稳定,但整体结构的变形会使其他长程相互作用变得不利 。例如,三聚体、四聚体或更大寡聚体之间的相互作用在这种情况下变得重要,同时晶胞尺寸的变化也会对能量产生影响 。这些寡聚体效应使得整体的势能阱比单个二聚体的势能阱更陡峭,并且晶体偏离中性应变越远,这些效应就越显著 。不过,这些效应仅在实验观察到的弹性变形极限之外才重要,在该极限之外可能会出现其他变形机制 。 (三)弹性恢复力的起源当[CuL21] 晶体弯曲到弹性极限(b 轴 1% 变形)时,晶体外侧的 [010] 轴膨胀,使得 π - π 相互作用能量增加0.29 kJ mol−1 ,同时 CH - π 和 CH - O 相互作用能量总共降低0.27 kJ mol−1 ,π - π 相互作用能量的变化比 CH - π 和 CH - O 相互作用能量的降低量多0.02 kJ mol−1 ,因此在晶体外侧,弹性能量主要存储在 π - π 相互作用中 。而在晶体内侧,[010] 轴被压缩,π - π 相互作用能量降低0.22 kJ mol−1 ,CH - π 和 CH - O 相互作用能量增加0.29kJ mol−1 ,导致0.07kJ mol−1 的弹性能量存储在 CH - π 和 CH - O 相互作用中 。所以,[CuL21] 弯曲环外侧的恢复力源于 π - π 相互作用,内侧的恢复力源于 CH - π 和 CH - O 相互作用的组合 。 在[CuL22] 和[CuL23] 中,π - π 相互作用在弯曲环外侧也起着类似的作用,而这两种材料中的其他相互作用相比[CuL21] 中的 CH - π 和 CH - O 相互作用影响力要小得多 。这表明在这一系列分子材料中,π - π 相互作用在拉伸应变下对恢复力的贡献具有一定的普遍性,但不同材料中其他相互作用的影响程度存在差异,这种差异与分子结构的变化密切相关 。 从能量存储的角度来看,在[CuL21] 晶体弯曲到弹性极限时,内侧存储的弹性能量约为70J mol−1 ,外侧约为20J mol−1 ,总存储弹性能量约为90J mol−1 ,[CuL22] 中也存储了类似数量的能量 。尽管与典型化学过程的能量相比,这是一个较小的量,但与许多机械能尺度相当 。例如,每摩尔[CuL21] 能够利用弯曲时分子间相互作用存储的能量将一个 9kg 的重物提升 1m,这表明这种分子材料在弹性变形过程中存储的能量具有一定的实际意义 。为了部分验证这一结果,研究人员制备了一系列单晶悬臂梁,将不同大小(最大可达晶体重量 100 倍)的钢球连接到[CuL21] 单晶上。施加力使钢球作用于晶体导致其弯曲,释放力后,钢球会被提升到比平衡位置更高的高度(补充视频 1 - 4),这一实验现象与理论计算得到的能量存储和恢复力的结论相符合。 (四)对塑性变形机制的启示研究结果还为[CuL21] 超出弹性极限后的塑性变形机制提供了进一步的见解。当晶体发生塑性变形时,平行于 (010) 面的平面会发生滑移,这需要破坏和重新形成 CH - π 相互作用 。由于 CH - π 相互作用的势阱相对较浅,相较于 π - 堆积相互作用更容易被破坏,所以这种滑移更容易发生,从而导致永久性变形。此外,在受力情况下,[CuL21] 晶体还会沿 [010] 方向发生纤维化,这也与 CH - π 和 CH - O 相互作用较弱的性质相符。因为这些弱相互作用在受力时难以维持晶体结构的稳定性,使得晶体更容易在这些方向上发生结构变化。这表明,分子间相互作用的特性不仅决定了材料的弹性行为,还对材料的塑性变形机制有着重要影响 。深入理解这些机制,有助于预测材料在不同受力条件下的行为,为材料的合理应用和性能优化提供指导。例如,在设计需要承受复杂应力的材料时,可以根据这些机制选择合适的分子结构,以提高材料的抗塑性变形能力 。 六、总体结论本研究通过对[CuL21] 、[CuL22] 和[CuL23] 三种分子材料的理论计算,成功地在原子尺度上确定了弹性恢复力的来源,揭示了分子材料弹性的起源。研究发现,在这些分子晶体中,不同的分子间相互作用在拉伸和压缩应变下分别主导恢复力 。在[CuL21] 中,拉伸应变时 π - π 相互作用起主要作用,压缩应变时 CH - π 和 CH - O 相互作用起主要作用 。这种相互作用的协同和竞争关系决定了材料的弹性行为,并且相互作用的 “受挫” 现象可能与材料的柔韧性相关 。 此外,研究还表明,分子结构的变化(如引入不同长度的脂肪链)会改变分子间相互作用的特性,进而影响材料的机械柔韧性 。通过对弹性变形过程中能量变化的分析,不仅明确了弹性恢复力的位置,还对材料的塑性变形机制有了更深入的理解 。这些发现为设计新型弹性材料提供了重要的理论依据,有助于推动柔性技术和智能设备的发展 。 从科研视角来看,本研究打破了以往对弹性恢复力仅作为宏观材料属性的认知局限,深入到原子尺度揭示其微观机制,为材料科学领域的理论研究开辟了新方向 。在未来的研究中,可以进一步结合实验手段,如高分辨率显微镜技术、原位力学测试等,对理论计算结果进行更直接的验证 。同时,拓展研究更多种类的分子材料,探索不同分子结构和相互作用对弹性性能的影响规律,将有助于开发出具有更优异弹性性能的材料 。例如,在柔性电子领域,可设计出在微小变形下仍能保持稳定电学性能的材料;在生物医学领域,开发与生物组织弹性相匹配的材料用于植入物或医疗器械 。总之,本研究为分子材料弹性的研究和应用奠定了坚实基础,具有重要的科学意义和应用价值 。 七、图文概览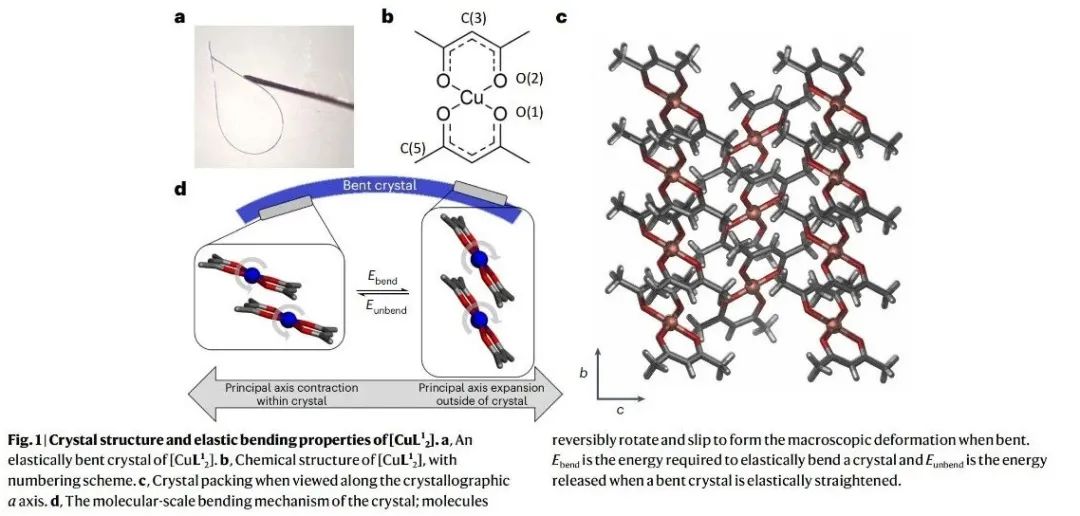 图 1、[CuL21] 的晶体结构和弹性弯曲特性。a、[CuL21] 的弹性弯曲晶体;b、[CuL21] 的化学结构及编号方案;c、沿晶体学 a 轴观察的晶体堆积;d、晶体的分子尺度弯曲机制,弯曲时分子可逆地旋转和滑移形成宏观变形。Ebend 是使晶体弹性弯曲所需的能量,Eunbend 是弯曲晶体弹性伸直时释放的能量。 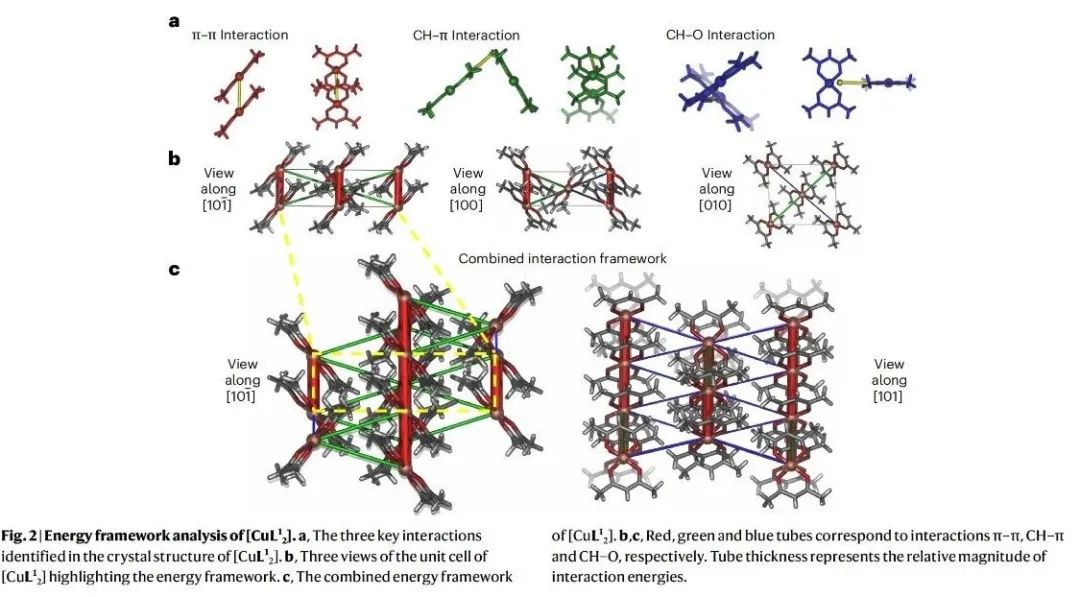 图 2、[CuL21] 的能量框架分析。a、在[CuL21] 晶体结构中确定的三种关键相互作用;b、[CuL21] 晶胞的三个视图,突出显示能量框架;c、[CuL21] 的组合能量框架。b、c 中,红色、绿色和蓝色管分别对应 π - π、CH - π 和 CH - O 相互作用,管的粗细代表相互作用能量的相对大小。 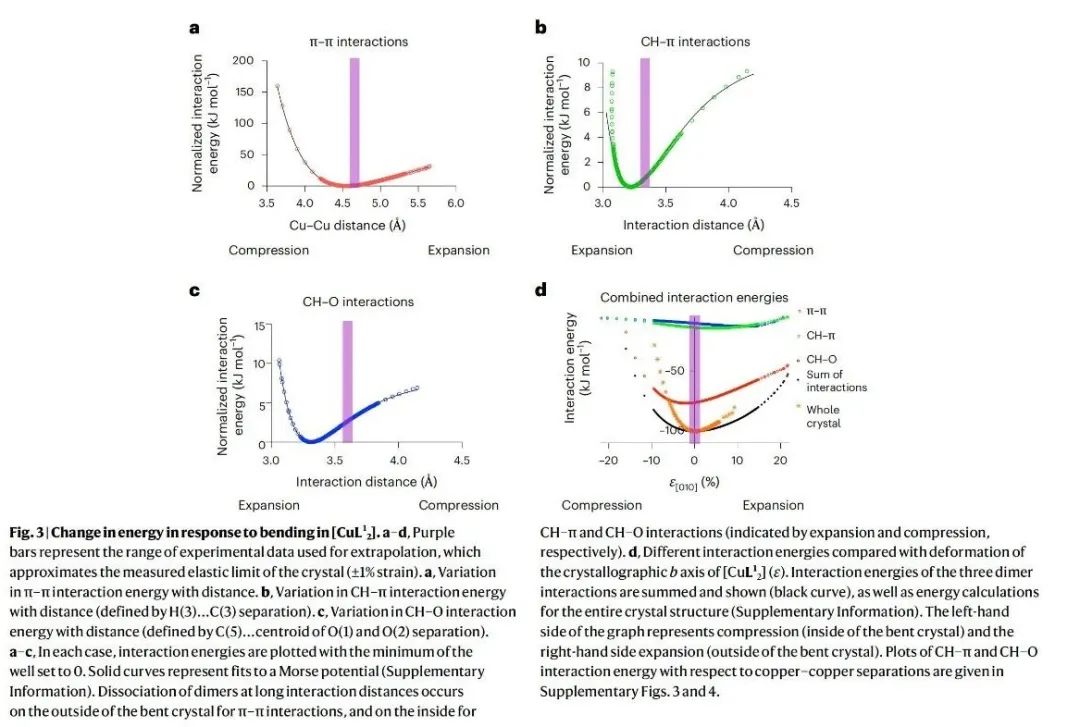 图 3、[CuL21] 弯曲时的能量变化。a - d、紫色条表示用于外推的实验数据范围,近似于晶体的测量弹性极限(±1% 应变)。a、π - π 相互作用能量随距离的变化;b、CH - π 相互作用能量随距离(由 H (3)…C (3) 间距定义)的变化;c、CH - O 相互作用能量随距离(由 C (5)…O (1) 和 O (2) 质心间距定义)的变化。a - c 中,每种情况下相互作用能量均以势阱最小值设为 0 绘制,实线表示对 Morse 势的拟合(补充信息)。在长相互作用距离下,π - π 相互作用的二聚体在弯曲晶体的外侧解离,CH - π 和 CH - O 相互作用的二聚体在弯曲晶体的内侧解离(分别由膨胀和压缩表示)。d、不同相互作用能量与[CuL21] 晶体学 b 轴变形(ε )的比较。三种二聚体相互作用的能量相加并显示(黑色曲线),同时也给出了整个晶体结构的能量计算(补充信息)。图的左侧表示压缩(弯曲晶体的内侧),右侧表示膨胀(弯曲晶体的外侧)。CH - π 和 CH - O 相互作用能量相对于铜 - 铜间距的图见补充图 3 和图 4。 八、作者信息作者姓名:Amy J. Thompson, Bowie S. K. Chong, Elise P. Kenny, Jack D. Evans, Joshua A. Powell, Mark A. Spackman, John C. McMurtrie*, Benjamin J. Powell*, Jack K. Clegg* 通讯作者及其单位: ·John C. McMurtrie,School of Chemistry and Physics and Centre for Materials Science, Queensland University of Technology, Brisbane, Queensland, Australia ·Benjamin J. Powell,School of Mathematics and Physics, The University of Queensland, Brisbane, Queensland, Australia ·Jack K. Clegg,School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Queensland, Australia 九、论文链接https://doi.org/10.1038/s41563-025-02133-w 十、版权声明本文来源各大出版社论文数据库,版权归文章出版社所有;本文内容采用 AI 辅助整理生成,如有错漏请私信联系;本文仅用于学术分享,转载请注明出处;如需推广本人学术成果请私信联系,若有侵权请私信联系删除或修改 |